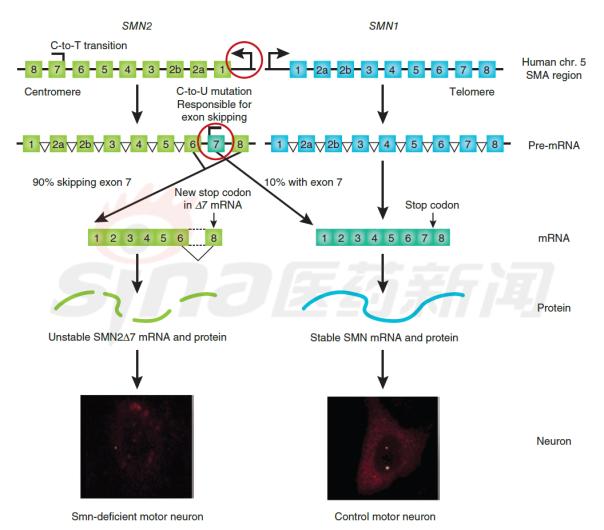
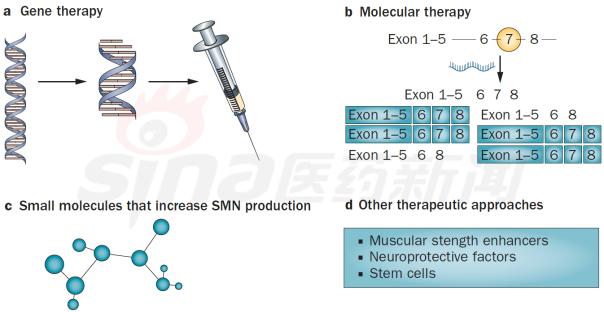
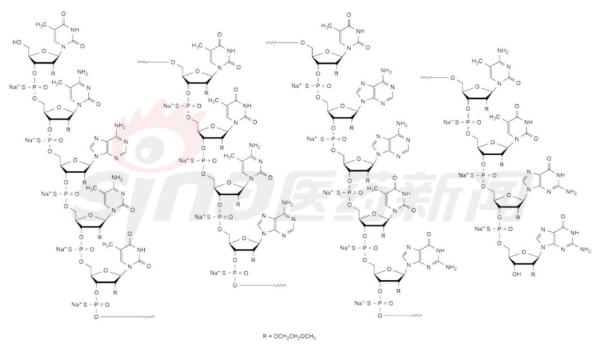
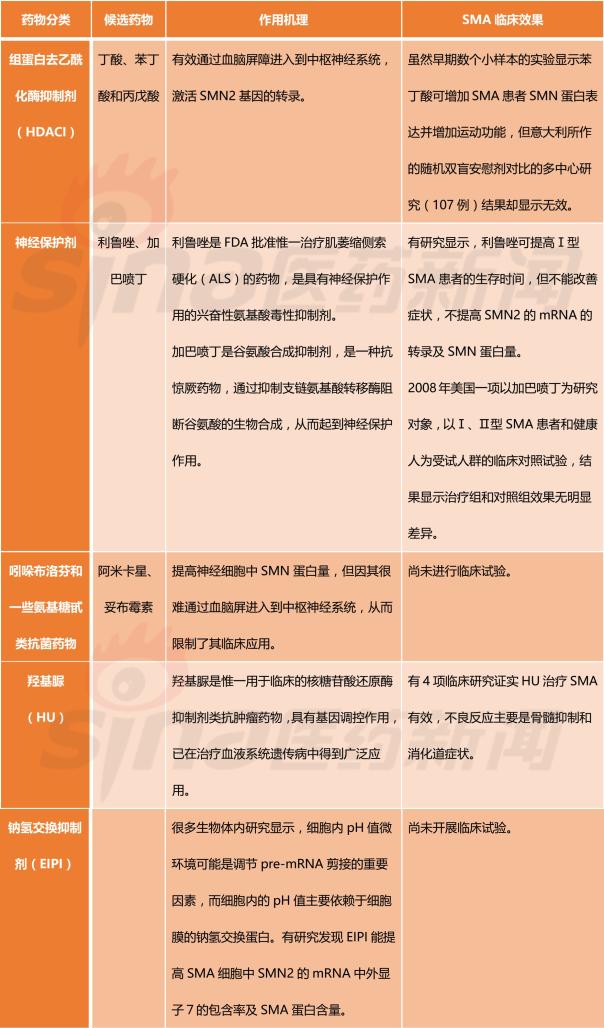
Focus on rare diseases: spinal muscular atrophy and its treatment progress January 16, 2018 Source: Sina medicine Author: unique eye medicine Spinal muscular atrophy (SMA) is a group of diseases in which genetic mutations cause cervical anterior horn cells to degenerate and cause clinical symptoms such as muscle weakness and muscle atrophy. According to the severity of the disease and the age of onset, the SAM International Symposium held at the European Neuromuscular Disease Center in 1992 divided it into four types (I, II, III, IV). According to statistics, the incidence of neonatal SMA is 1/6000~1/10000, ranking second in the fatal autosomal genetic disease, second only to cystic fibrosis. SMA is caused by the variation of motor neuron survival gene (SMN). With the study of the characteristics of SMN and the function of its encoded protein (SMN protein), clinical trials of various therapeutic strategies are advancing. 1 , clinical symptoms and classification SMA is mainly characterized by progressive, symmetrical limb proximal and trunk muscle weakness and atrophy. The patient eventually died of respiratory failure and severe pulmonary infection. SMA is classified into 4 types according to the age of onset of the patient and the severity of the disease. As shown in Table 1. SMA-I is the most common subtype, accounting for about 50%, with acute onset and severe illness. Patients usually die within 2 years of age; children with type II SMA are slightly milder than type I, but children There are still difficulties in sitting, eating, breathing, etc. Type III patients have a wide range of onset, as early as 1 year old, and may develop from late to adolescence. Generally, type III patients have normal early exercise indexes, but they gradually show signs of decline in motor function with age. The symptoms of type IV patients usually start after 35 years old, which is rarer than other types. The typical feature is invisible disease. Development is very slow. Table 1 Basic conditions of each subtype of SMA 2 , molecular genetics and etiology The motor neuron survival gene (SMN) is located at 5q11.2–q13.3 and is responsible for encoding the SMN protein. If the SMN is mutated, the SMN protein content and activity are reduced, and the spinal cord anterior horn cells are intolerable and a large number of apoptosis, resulting in SMA. disease. The SMN gene has two highly homologous copies: SMN1 and SMN2. The main functional difference between SMN1 and SMN2 is exon 7. SMN1 encodes a full-length SMN transcript (SMN-fl), while SMN2 encodes a large number of selective transcripts of skipped exon 7 (SMNΔ7) and a small amount of SMN-fl. SMN-fl contains exon 7 and SMNΔ7 does not contain exon 7. Exon 7 is essential for the production of a stable, fully functional SMN protein. Thus, SMN1 encodes a fully functional SMN protein, and SMN2 alone does not provide sufficient full-featured SMN protein to maintain the survival of human motor neurons. The SMN protein contains 294 amino acids with a molecular weight of 38 kD. It is widely distributed in whole body tissues and has different expression levels, with the highest expression levels in brain, spinal cord and muscle. When the SMN gene is deleted or mutated, the number, structure and function of the expressed product SMN protein are abnormal. Motor neuron degeneration occurs when the amount of SMN protein is lower than the minimum required for motor neurons. Figure 1 Schematic diagram of the functions of genes SMN1 and SMN2 3 , clinical diagnosis The main clinical manifestations of SMA are progressive, symmetrical limb proximal and trunk muscle weakness, atrophy. For patients with suspected SMA, some tests are needed, including creatine kinase quantification, electrophysiologic alanalyses, imaging studies (MRI), and specific genetic and metabolic tests. Assessment). The most common mutation in SMA patients is the homozygous deletion of the SMN1 gene. Most carriers can detect the deletion of one allele in the SMN1 gene and detect the exon 7 and 8 of the SMN1 gene, which is the diagnosis of SMA gene. The preferred method of prenatal diagnosis. SMA is one of the common hereditary diseases, with a carrier frequency of about 1 in 50. Conducting carrier genetic screening and necessary prenatal diagnosis is a recognized preventive measure. Couples who have had children with SAM, the risk of recurrence in children with SMA is 25%, and prenatal diagnosis is required during each subsequent pregnancy. 4 , SMA treatment Patients with SMA lack SMN protein, so the ultimate goal of treatment is to increase the amount of SMN protein. Based on this goal, strategies can be adopted to: replace or modify the variant SMN1 gene; correct negative/positive regulation during gene splicing to promote transcription of exon 7 in the SMN2 gene; increase the activity of the SMN2 gene transcription promoter; protect SMN Protein and SMNΔ7 protein and improve their stability. The main treatments currently under investigation include: Genetherapy, Antisense oligonucleotides (ASOs), Small molecules, and some adjuvant treatments (stem cells, muscle-enhanced therapy, nerves). Protection treatment). as shown in picture 2. Figure 2 Current SMA treatment strategy Gene therapy Currently the only gene therapy project to enter the clinical stage is AVXS-101 from AveXis, Inc. AVXS-101 has obtained the orphan drug qualification for the treatment of SMA, and has obtained the breakthrough therapy identification and rapid review channel for the treatment of SMA-I. The therapy uses a serotype 9 adeno-associated serotype 9 virus (AAV9) as a vector to introduce a human SMN gene into motor neuron cells. Because AAV9 is able to cross the blood-brain barrier, AVXS-101 can be administered intrathecally or intravenously. The introduced human full-function SMN gene is a self-complementary (sc) double-stranded molecule, which skips the general single-stranded genetic material virus and requires the host cell to regulate the synthesis of the double-strand. The SMN protein is encoded and works quickly. In addition, AVXS-101 also introduced a well-designed hybrid cytomegalovirus control enhancer/chicken-β-actin promoter to ensure that the SMN gene continues to stably encode SMN proteins. To investigate the safety and tolerability of AVXS-101, Phase I clinical trials involving 9 SMA patients (0-9 months) are ongoing and no relevant results have been disclosed. A potential concern of the project is that although intravascular injection can reduce the amount of viral vector used, the introduction of the SMN gene into the nervous system still requires a large number of viral vectors. Antisense oligonucleotide drug Indirect rearrangement of abnormal mRNAs using small RNAs is the focus of an extended research area of ​​common genetic diseases, including SMA. Modulation of SMN2 mRNA restores the expression of the appropriate SMN protein for the purpose of improving the symptoms of SMA patients. Research in the field has focused on antisense oligonucleotides (ASOs). A specific sequence exists on SMN2 that regulates the SMN2 encoding deletion of mRNA for exon 7. ASOs bind directly to this specific sequence and inhibit its regulation, prompting SMN2 to encode a fully functional SMN protein. In an experiment, according to the structural characteristics of the SMN2 gene, the AOSs were spliced ​​and combined into a bifunctional RNA group, and the bifunctional RNA groups were injected into the lateral cerebral ventricle of the mouse model to increase the content of SMN protein in the brain. Some preclinical studies have shown that 2'-O-methoxyethyl (MOE) and morpholinyl ASOs can bind tightly to the SIN2 gene's own splicing silence sequence N1 (ISS-N1). Tests have shown that both ASOs can alleviate the symptoms associated with SMA model mice. It is worth mentioning that Isis Pharmaceuticals (Carlsbad, CA, USA) has introduced the MOE-ASO drug candidate ISIS-SMNRx to the clinic, and Phase I and II trials have confirmed its safety and potential efficacy. Two phase III clinical trials, ENDEAR and CHERISH, also achieved satisfactory results. In December 2016, the drug was approved by the FDA, under the trade name Nusinersen, becoming the first listed drug to treat SMA. Nusinersen is administered intrathecally, once every four months, at a dose of 12 mg (5 mL). The main side effects of the drug are: thrombocytopenia, abnormal blood clotting and kidney toxicity. The molecular formula of Nusinersen is C234H323N61O128P17S17Na17, and the molecular weight is 7501.0. Its chemical structure is shown in Figure 3. The advantages of ASOs treatment strategy are simple and clear product structure, good safety, obvious therapeutic effect, specific effect and less side effects. At the same time, ASOs and SMN2 genes are reversible and do not introduce foreign genes. The current problem with ASOs is that repeated dosing is required, and the biodistribution of the drug in the nervous system and its efficacy are not balanced. Figure 3 Nusinersen molecular structure Small molecule drug Over the past few years, a series of small molecule compounds that increase SMN protein have been identified by high-throughput screening methods that increase the activity and longevity of neuronal cells in SMA model mice. The small molecules currently under study are shown in Table 2. The advantage of small molecule drugs is that the structure is simple and convenient for oral administration. The disadvantage is that the specificity is not strong and the side effects are serious. Table 2 Summary of small molecule candidate drugs Plastic Security Seals,Plastic Security Seal,plastic seals,plastic seal Wenzhou Haoshi Light Industrial Products Co., LTD , https://www.economicseals.com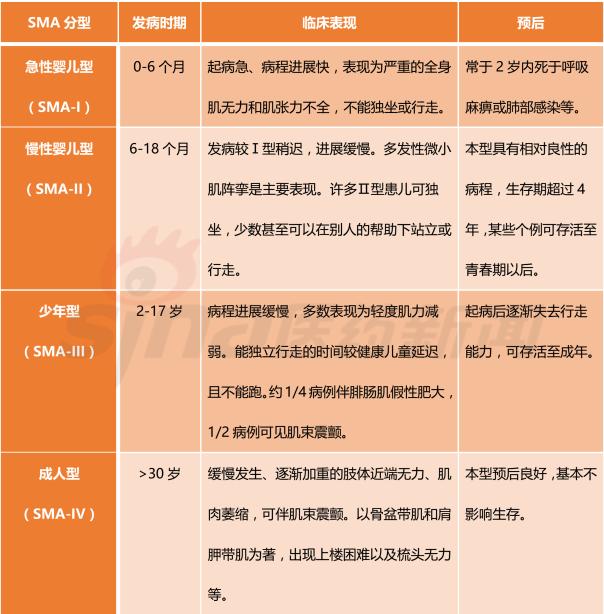
Focus on rare diseases: spinal muscular atrophy and its treatment progress